Within all of our cells, we have millions of proteins, nucleic acids, lipids, and carbohydrates serving various biological functions. Many of these molecules have structure which play key roles in understanding how the mechanisms of those biological functions work. Scientists who study structural biology seek to determine the structure of these important players, in order to better understand how our cells work, how dysregulation occurs, and in a medical setting, how we can inhibit such dysregulation.
The Basics of Protein Structure
Protein structure is divided into four main categories: primary, secondary, tertiary, and quaternary. Primary protein structure is the sequence of amino acids constituting the peptide chain. Each of these amino acids have their own individual structures that affect their properties (electronegativity, polarity, size, hydrophobicity, etc) and how they interact with one another (hydrogen bonds and van der waals forces, for instance). For further explanation on those proprieties and the structure of each, see this blog post discussing properties and this one for helpful tips on memorizing their structures.
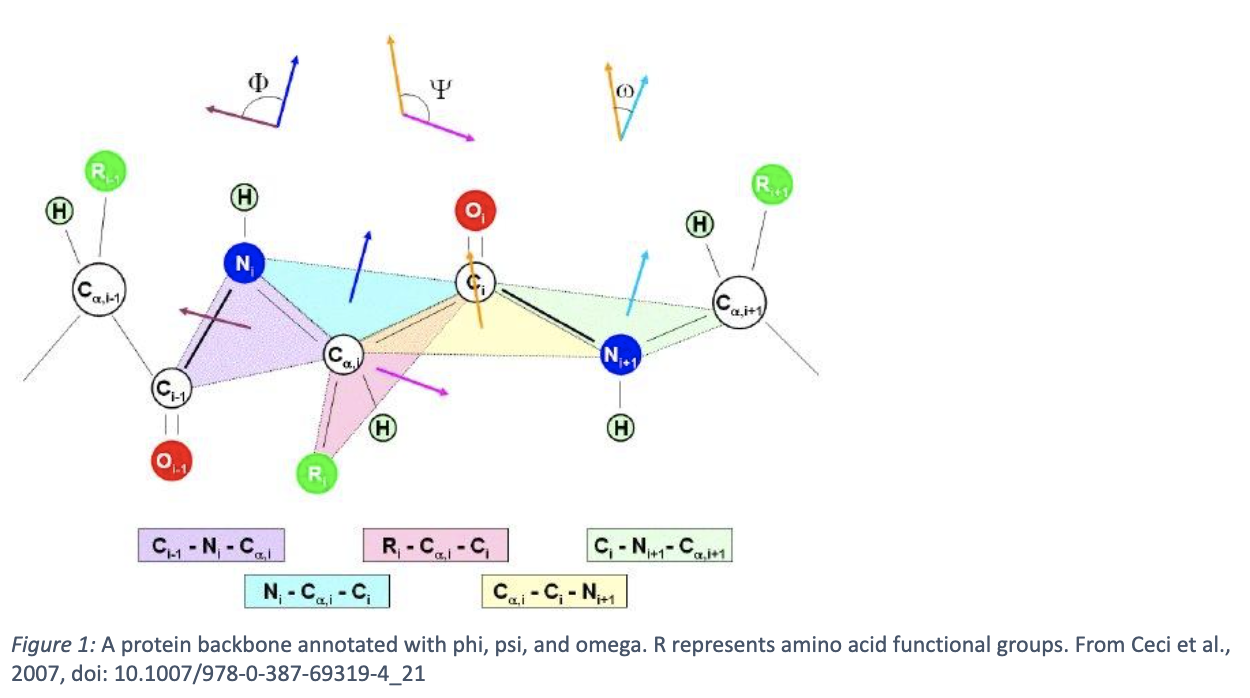
Next is secondary structure. Due to the aforementioned properties of amino acids and their interactions with each other, the peptide chain can fold into specific conformations. Most commonly, these are α-helices and β-sheets, although less common structures such as other loops, turns, and alternative helices, exist as well. These each have well characterized geometric properties, like pitch for helices and parallelity of strands in β-sheets. The geometry of the peptide backbone can be described by dihedral angles: phi, psi, and omega, shown in figure one below. The allowed dihedral angles for any given residue can be described by a Ramachandran plot, which is a heatmap describing how likely it is for that given residue to adopt a given angle between -180 and 180 degrees. Certain angle pairs suggest what secondary structure that amino acid is a part of (see figure two).

According to these structural properties, the protein adopts a tertiary structure. Here, it gets complicated – it’s very difficult to predict the three-dimensional structure of a protein from its peptide sequence. Advanced ML algorithms like RoseTTAFold and AlphaFold have recently tackled this problem, but there is still much error in complex proteins, multi-domain enzymes, protein binding, and multi-state proteins. Furthermore, understanding folding from first principles remains elusive – see the protein folding problem for more information. However, what we do know is that a protein seeks to minimize its Gibbs Free Energy by folding. One instance of this is by burying hydrophobic residues within the protein and exposing hydrophilic residues, which “frees” the surrounding water molecules, thus increasing system entropy even while the entropy of the protein decreases. The protein forms further intermolecular bonds, such as disulfide bonds, hydrogen bonds, and ionic interactions, inducing further stability to the molecule. While the protein structure is “encoded” by its sequence, it cannot always fold natively in solution, and sometimes requires a chaperone. Further, because there can be multiple energetically favorable conformations, a protein structure can dynamically change. Finally, those tertiary structures can be combined into a full, multi-subunit protein complex, based on further energetically favorable interactions – this is termed the “quaternary” structure of a protein.
The Basics of Nucleic Acid Structure
Analogously to protein structure, nucleic acids also fold into specific structures – particularly, single-stranded nucleic acid structures adopt a variety of biologically important secondary and tertiary structures. The primary structure for nucleic acids is the sequence of the (canonical) four nucleic acids for DNA (adenine, cytosine, guanine, and thymine) and the four for RNA (the same but replacing thymine with uracil). Noncanonical nucleic acids also exist and are particularly important in RNA structure.
To understand secondary structure in nucleic acids, it is important to understand that adenine pairs with thymine/uracil, and cytosine with guanine, in what is referred to as “Watson-Crick base pairing.” Adenine and guanine are referred to as “purines” because of their similar double-ringed structures, while uracil, thymine, and cytosines are grouped as “pyrimidines” due to their single aromatic ring. Noncanonical nucleic acids also follow base pair rules. Double-stranded DNA usually follows these binding rules to form the well-known double-helical structure. Because RNA usually exists as a single-stranded molecule, it can adopt a more diverse array of conformations, such as hairpins, pseudoknots (intertwined hairpins), and bulges, that are commonly seen in functional RNAs and viral RNA genomes.
Similar to amino acid influence on protein structure, nucleotide sequence affects nucleic acid tertiary structure. The most common conformation of dsDNA, B-DNA, can adopt different helix pitch in response to the stacking forces that each nucleotide exerts on its neighbors. The stability of the helix itself is determined by ratio of A-T and G-C bonds, which have 2 and 3 hydrogen bonds respectively. A-DNA has the same handedness as B-DNA, but a much more compact structure, usually forming under dehydrating conditions. Long purine-pyrimidine repeats can even cause different handedness; Z-DNA, which is named because of its zig-zag looking structure, is right handed instead of left handed. RNA tertiary structure comes when the secondary structural elements fold into sometimes stable, and sometimes unstable, three-dimensional configurations.
Analogously again to protein structure, quaternary structure occurs when multiple molecules come together. Importantly, DNA-protein and RNA-protein structures exist, as well in quaternary structures, providing important biological functions such as DNA packing and protein translation.
How do researchers determine structures?
Because all these aforementioned structures are on the scale of angstrom-to-nanometer size, it is nontrivial to determine structures of biological molecules. Three main techniques are used to determine structures: x-ray crystallography, cryo-electron microscopy (cryo-EM), and nuclear magnetic resonance (NMR).
X-ray crystallography is the classic technique to use, where crystals of proteins or other biological molecules are formed and grown, and then x-rays are diffracted off of them, giving a diffraction pattern which can be used to determine structure (see the famous diffraction pattern, taken by Rosalind Franklin, of DNA). It is particularly useful for small, well-structured proteins, although large structures have been solved as well.
Cryo-EM has risen to prominence in the past decade, and relies on electron microscopy, in which electrons are scattered off of a biological sample and detected on a camera to form an image. Many repeating images in different conformations can be used to reconstruct a three-dimensional image from those two-dimensional signals. It is useful for large structures and structures with a high degree of symmetry.
In NMR, biological molecules are put into a large magnetic field, then subjected to an oscillating magnetic field at resonance. Nearby atoms affect the resonant frequency, and so by understanding how each atom differs from a characteristic resonance, scientists can solve the structure of a molecule by knowing what nearby atoms exist to a given atom. Because molecules are in solution during NMR, it is particularly useful for small, dynamic molecules.
Why should I care?
You might be wondering why structural biology matters – why is it on the MCAT, and why do billions of dollars get spent every year towards solving structures? There are a few reasons. For one, structural biology gives us more complete insight into and understanding of molecular mechanisms within our cells – it helps us understand how things work. This has a few benefits. For one, some molecular machines are incredibly powerful – for instance, the type IV pilus machine can pull 100 pN per pilus, or 10,000 times the weight of a bacteria (assuming 10 fN). This has implications for intelligent design. Further, vaccines are increasingly being developed using rational drug design – designing specific molecules to inhibit the entry of foreign agents. This is based upon structural analysis. Thus, understanding structural biology not only helps one understand the basic inner workings of our biology, but also has practical applications in synthetic biology and biomedical sciences.


Comments